GCG Workshop, October 2003
Emphasis on SeqLab Interface
Author: Thomas Girke (thomas.girke@ucr.edu)
Contents
B. Starting SeqLab and Command-Line GCG
C. Using the Working Directory
D. Working with the Main SeqLab Window
E. Editing and Annotating Sequences
F. Importing and Exporting of Sequences
Database Searches: BLAST, NetBLAST, PsiBLAST, HMMER, etc.
Alignments: Pileup
Pattern Finding: MEME, Motifs, FindPattern
Protein Analysis
Useful Tools: FrameSearch, FrameAlign, etc.
K. Large-Scale Sequence Analysis: BLAST Example
N. Creating Personal Sequence Databases with DataSet
The Wisconsin Package GCG is a software suite that contains over 130 sequence analysis tools. It was developed by the Genetics Computer Group in Madison and is now maintained and distributed by Accelrys. It can be accessed remotely from any networked computer. There are three different interfaces for accessing GCG:
Command-Line
SeqLab: graphical X-windows interface
SeqWeb: web browser interface (http://gcg.ucr.edu)
All three applications are installed on the UNIX server cache.ucr.edu where they share the same sequence databases. The instructions for setting up an account can be found on our GCG page. This workshop will focus on SeqLab, since it is the most powerful and versatile GCG interface.
To run SeqLab from a PC, you need to configure X-win32 (for Mac OS X: X11, configuration) and PuTTY according to the configuration page. For transfering files between your local machine and the GCG server, I recommend using WinSCP (for Mac OS X: Fugu). More help on configuration issues can be found on our GCG page.
B. Starting SeqLab and Command-Line GCG
SeqLab:
Start X-Win 32
Log into cache.ucr.edu (chug.ucr.edu) using PuTTY (X11 in Mac OS X)
$ seqlab & ("&" starts it in background)
Start GCG command-line (can be in addition to SeqLab):
$ gcg
Help: In SeqLab you find help documents by clicking on the Help menu in the window of the different applications. On the command-line you can open these help files with the command "genhelp" or "genmanual". To retrieve help for specific programs, just type its name ofter these commands. Additional information can be found in the (Online GCG Manual, usr: genhelp, pwd: version102). General help on UNIX can be found on the same page under User's Guide.
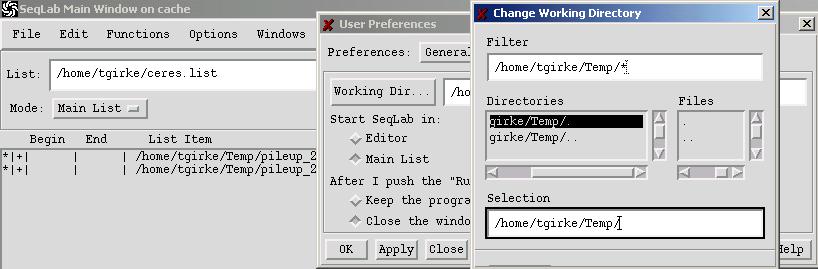
C. Using the Working Directory
The Working Directory window is one of the most important components of SeqLab. In this window you specify the directory where SeqLab writes output files. Remember, in GCG you generally create in each session many output files. Not using this feature can create a big mess in your account.
To access this function, go to:
Options -> Preferences -> Working Directory
Navigate to the directory you want to use by typing in its path in the Filter window and then hit enter. A double-click on the two dots ".." in the Directories field brings you to the next higher directory.
New directories can be created by typing in their name in the Selection text box and then clicking OK. A convenient tool to create and manage new directories is WinSCP.
SeqWeb users can copy their files on the command-line from /usr/local/seqweb/2.0.2/seqweb/html/user/your_account_name/work/ into their home directory.
D. Working with the Main SeqLab Window
Main List
The Main List window is SeqLab's project managing tool that allows you to organize data on a project-by-project basis. Here and in the Editor (s. below) you select the sequences you want to analyze with the different tools which are available in GCG. You can switch between the Editor and Main List in the Mode menu (3).
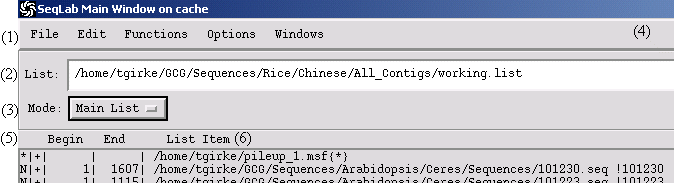
Menu Bar:
File: load and save projects
Edit: edit items in Main List and Editor
Functions: select GCG software tools
Options: working directory and graphics devices
Windows: access job management, trace file view and sequence features
List: currently loaded list file. It has nothing to do with the working directory!!!!!!!!!
Mode: switches between Editor and Main List
Help
Attributes: nucleic acid (N), protein (P), unknown (*), forward (+), reverse (-), length, etc.
List Item: File names (path)
Editor
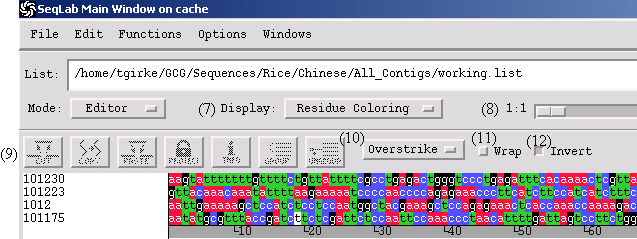
Display: select between different color modes
1:1: Zoom
Icon Bar: Cut (excises and copies selected area), Copy, Paste, Protect (sets protections), Info (displays information on sequence)
Options: Overstrike (replaces & and deletes at cursor), Insert (inserts and deletes at cursor) and Check (retype check with beep warning)
Wrap: switch between wrapped and unwrapped display
Invert: switch background and character coloring
Navigation Bar: indicates position, column, orientation, etc.
E. Editing and Annotating Sequences
Transformations
The common sequence editing and search functions can be found in the Edit menu:
select the sequence files or sequence areas you want
go into the Edit menu and select Reverse, Find, Translate, etc.
A nice feature of SeqLab is that it allows you to perform these operations on many sequences at once instead of doing it on a one-by-one basis as it is the case in most other sequence editors.
Annotations
To add annotations to a sequence or alignment you can do this within the sequences (see Sequence Features) or in a separate comment line. To add a comment line, you select in the Editor window File -> New Sequence -> Text. A new line appears which can be moved under the sequence of your choice by using the copy and paste buttons. Switch to Insert mode and add your comments. All changes can be saved in RSF format.
Note: To create and edit sequences from the command-line, you can use SeqEd which is an additional interactive sequence editor in GCG.
F. Importing and Exporting Sequences
Import
There are three major ways to import sequences into GCG:
One-by-one import:
Switch in main window to Edit mode
File -> Import -> Select sequence and click OK -> Specify type of sequence.
To save sequence in GCG format, select the sequence in Edit mode -> File -> Save As -> <name.seq>
Import of MSF alignments (FASTA formatted alignments can be imported via batch import):
Switch in main window to Main List mode
File -> Add Sequences From ->Sequence Files -> Select alignment and click OK.
Batch import (imports single sequences as well):
To import many sequences in a single operation, they need to be in one concatenated file. If you have them in separate files, you can combine them with the "cat" function on the command line: $ cat *.seq > batch.seq
Functions -> Importing/Exporting -> <select format> -> browse to your file and give new list file a name such as *.list
The
GCG formatted sequences are saved as separate files into your
working directory. Each file receives the name of its sequence ID,
which is in a FastA file the text behind the ">".
If your sequences are in a different format, you can reformat them on the command line:
foreach f (*.seq)
? chopup $f -def
? end
reformat *.dat
Export
There are two possibilities to export sequences from GCG:
Sequences and alignments that were modified in the Editor can be exported into MSF or GenBank format by selecting them in the Editor and choosing File -> Export -> <select format>.
To export sequences to FastA and Staden format, you select the sequences in the Main List and choose Functions -> Importing/Exporting -> <select format>. When you select FastA as output format you have the choice (under Options) to export each sequence to a separate file or into one FastA batch file. The latter one is often preferred if you want to import your files later into other databases.
G. Trace Files, Assembly and Mapping
Import of trace files:
switch to Editor
Import trace files (ABI and SCF format)
Select the sequence files in Editor -> from Windows menu choose Traces. The sequences can be edited in the Editor and the changes will appear in the Trace Viewer.
Changes can be saved in rsf format (rich sequence files), which contains the edited sequences and trace information in one file.
Assembly
Due to time restrictions, the workshop will provide only a brief summary of the different sequence assembly tools which are available in GCG.
Assemble: concatenates sequence files in input order
The "Gel..." tools are linked together and need to be used in the specified sequence:
GelStart: creates a new fragment assembly project
GelEnter: adds specified sequences to assembly project
GelMerge: assembles sequences in assembly project into contigs
GelAssemble: lets you view and edit the contigs assembled by GelMerge:
select FAS in GelAssemble window
up and down key to select contig, CTRL&K to load contig
move curser with arrow keys and type in changes
to save changes, switch with CTRL&D to command mode, type in WRite or Exit and hit enter.
For details read GelAssemble Help/Command Mode
GelView: displays structure of contigs in assembly project
GelDisassemble: breaks up all contigs into their original fragments
Mapping
Prime selects oligonucleotide primers for a template DNA sequence. You can allow it to use for the design the entire template or use a list of your primers.
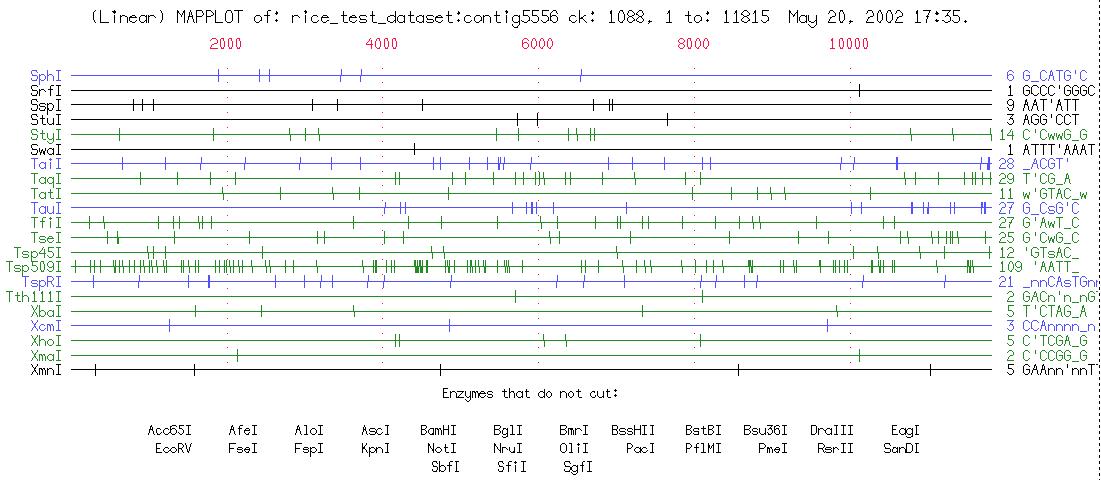
Map displays enzyme restriction sites above both strands of DNA along with protein translations below the DNA (see fig. below).
MapPlot displays restriction sites graphically.
MapSort lists, by size, the fragments of single or multiple restriction enzyme digests.
PlasmidMap reads file from MapSort (run with the command-line parameter -PLAsmid) to draw plasmid maps.

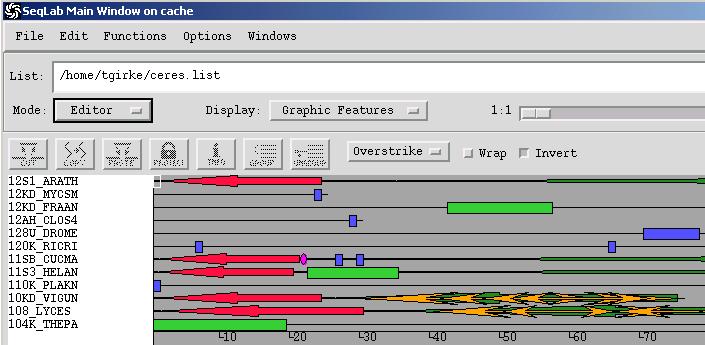
View Features
Annotation features such as introns, domains and structural information from public and personal databases can be graphically displayed in the Edit window by choosing in the Display menu the options Features Coloring or Graphics Features.
To display features from unaligned sequences in a Pileup alignment, do the following from the Editor:
Load the annotated sequences into the Editor
Create a multiple alignment with Pileup
Add the newly created MSF file from the Output Manager to the Editor. When prompted, choose to "overwrite the existing sequences". Your alignment will be loaded in the editor, and the annotation from the unaligned sequences will be properly carried over.
Edit Features
Highlight sequence file or sequence area in the Edit window, then choose from the Windows menu the option Features and provide in the resulting window your annotation information. Graphical symbols can be chosen in the sub-windows Edit and Add.
For advanced users:
Features can be customized in the file feature.cols, which needs to be localized in the directory from where you start SeqLab (/home/user/). To move this file there, type on the command-line $ fetch feature.cols. Use your favorite editor to modify this file according to your preferences.
The easiest way to print graphics or integrate them into other graphical applications, is to save them in PostScript format and to transfer the resulting file to your local computer, where you can modify and print it in Ghostview, a free software which can be downloaded from this page: http://www.cs.wisc.edu/~ghost/index.htm. When you do this for the first time you have to enable the PostScript format in SeqLab under Options -> Graphics Devices -> Language: PostScript
a) To print sequences and alignments to a file:
view them in the Editor -> File -> Print
in the Print window select PostScript in the field Output Format and File in the field Destination.
b) To print graphics from other GCG applications such as PepPlot:
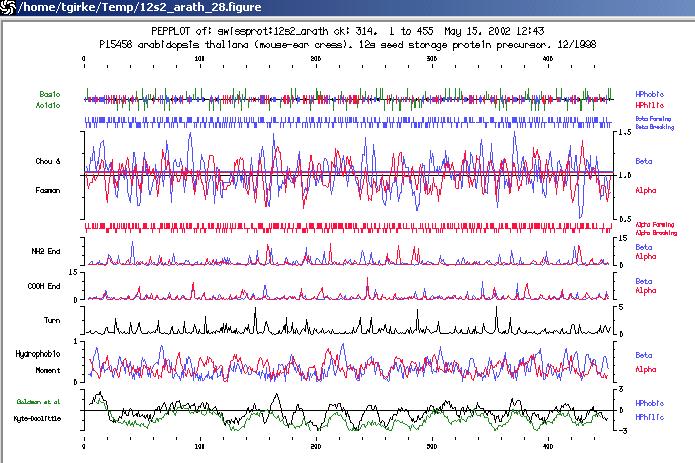
click on Print at left corner of this window
type in a file name in the field Port or File.
Most GCG programs can be accessed through the option Functions in the Menu Bar of the main window, which provides access to currently 111 different sequence analysis tools. This workshop can provide only a brief introduction into a small selection of this massive collection of GCG programs.
For an efficient job and database management, please make yourself familiar with the following functions in the Windows menu: Job Manager, Output Manager and Database Browser.
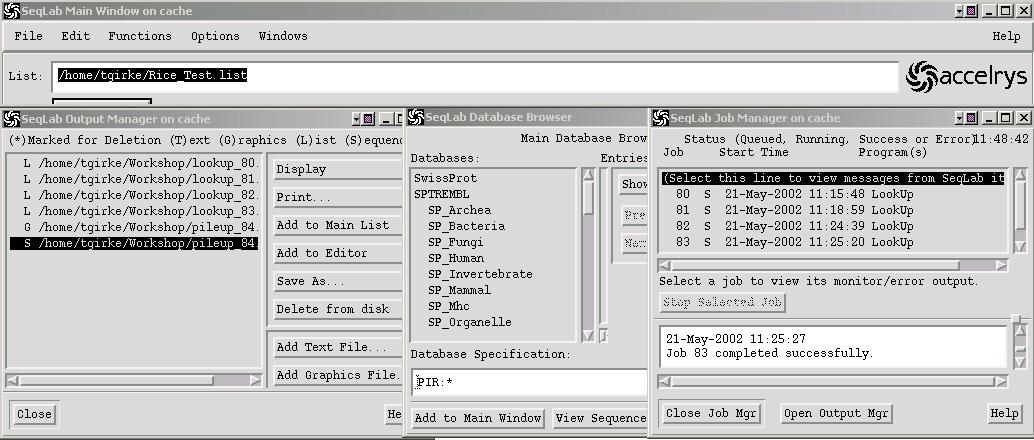
Database Searches
- Lookup identifies sequence database entries by name, accession number, author, organism, keyword, title, reference, feature, definition, length, or date. The output is a list file of sequences, which can be used to load all specified sequences into the Main List or Editor.
- BLAST searches local nucleic acid or protein databases. This important function will be introduced in the next paragraph (K).
- NetBLAST searches NCBI's database online.
- FastA does a Pearson and Lipman search for similarity between a query sequence and a group of sequences of the same type. For nucleotide searches, FastA may be more sensitive than BLAST.
- SSearch does a rigorous Smith-Waterman search for similarity between a query sequence and a group of sequences of the same type (nucleic acid or protein). This may be the most sensitive method available for similarity searches. Compared to BLAST and FastA, it can be very slow.
- PSI-BLAST: Position specific iterative BLAST (PSI-BLAST) refers to a feature of BLAST in which a profile (or position specific scoring matrix, PSSM) is constructed (automatically) from a multiple alignment of the highest scoring hits in an initial BLAST search. The PSSM is generated by calculating position-specific scores for each position in the alignment. Highly conserved positions receive high scores and weakly conserved positions receive scores near zero. The profile is used to perform a second (etc.) BLAST search and the results of each "iteration" used to refine the profile. This iterative searching strategy results in increased sensitivity.
- HMMER can be used to perform sensitive database searching using statistical descriptions of a sequence family's consensus. Related software packages are PSI-BLAST and SAM. A very nice user guide on HMMER can be found on Sean Eddy's home page (http://hmmer.wustl.edu/).
HmmerAlign aligns multiple sequences to a profile HMM. It can be used to create alignments of large numbers of sequences. HmmerBuild builds a profile HMM from a given multiple sequence alignment. HmmerCalibrate determines appropriate statistical significance parameters for a profile HMM prior to doing database searches. HmmerConvert converts HMMER profile HMMs to other formats. HmmerEmit generates sequences probabilistically from a profile HMM. HmmerPfam searches a profile HMM database with a sequence. HmmerSearch searches a sequence database with a profile HMM.
Alignments (Example)
- Pileup creates a multiple alignment of unaligned sequences. The alignment is written to a MSF file which can be imported into many alignment editing tools, such as GeneDoc.
Pattern Finding
- MEME finds conserved motifs in a group of unaligned sequences.
- Motifs looks for sequence motifs by searching through proteins for patterns defined by PROSITE.
- FindPatterns looks for patterns defined by the user.
Protein Analysis: Browse through the different protein analysis tools to identify which ones may be useful for your work.
Example:
- PeptideStructure makes secondary structure predictions including alpha, beta, coil, turn, antigenicity, flexibility, hydrophobicity and surface probability. A very useful exercise on predicting structure and antigenicity of peptides can be found on this page: http://mcf.ahc.umn.edu/Tutorials.htmls/minitutor6.html
Useful Tools
- FrameSearch searches a group of protein sequences for similarity to one or more nucleotide query sequences, or searches a group of nucleotide sequences for similarity to one or more protein query sequences. For each sequence comparison, the program finds an optimal alignment between the protein sequence and all possible codons on each strand of the nucleotide sequence. Optimal alignments may include reading frame shifts.
- FrameAlign creates an optimal alignment of the best segment of similarity (local alignment) between a protein sequence and the codons in all possible reading frames on a single strand of a nucleotide sequence. Optimal alignments may include reading frame shifts.
- BackTranslate backtranslates an amino acid sequence into a nucleotide sequence. The output helps you identify areas with fewer ambiguities that might be candidates for synthetic probes.
K. Large-scale Sequence Analysis: BLAST Example
Many sequence analyses in GCG can be performed in a batch pipeline. The sequence search tools FASTA and BLAST are just two of many of those "batch" applications, which query sequences databases that are installed locally on cache.ucr.edu. The application NetBLAST allows you to perform online searches against sequence databases on the NCBI server, but it is limited to one sequence submission at a time.
To run many BLAST and FASTA searches at once on cache.ucr.edu, you must first select the sequences of your choice in the Main List or Editor. For selecting sequences you have several options:
Select individual sequences in Main List or Editor
Select a database or DataSet (see below) in Main List or Editor
Select a list file of "sequence pointers" the Main List or Editor
To start the BLAST search with the selected sequences, you choose Functions -> Database Sequence Searching -> BLAST. In the open BLAST window you need to select whether you want to search a nucleotide/protein database (defines use of BLASTN, BLASTX, TBLASTN, TBLASTX) and the Search Set (specifies database). Under Options you set the search parameters:
here you normally want to reduce the number of sequence hits from the default of 500 (which is a waste of storage space) to a much smaller number
under Format for Alignments you have the choice between many options: standard, XML, tab delimited, etc. Some of these options can often simplify the downstream data parsing.
usage of filters and masks
scoring matrix: default is BLOSUM62, you have the option to change to BLOSUM45, 80, and PAM30, 70 by clicking on Specify Scoring Matrix
many other parameters...
Note: When you perform batch operations in GCG, the software names the output after the sequence/query ID#s and their file extensions correspond to the name of the search tool. Example: gi343848.tblastx.
For parsing of BLAST result, you can try to use on the command line our Perl script "blastParse" or this simple Perl one-liner:
perl -ne 'print if (/Query=/ ? ($c=1) : (--$c > 0)) ; print if (/End of List/ ? ($d = 9) : (--$d > 0))' input.blast > output.parse
List files are a very efficient way to perform analyses of specific sets of sequences. Since they contain only pointers to the sequences, they can save you a lot of storage space (no duplication of large sequence data) and allow very quick selections of defined sequence groups to perform various analyses simultaneously. For instance, one can quickly create a list file for thousands of sequences in a spread sheet program and submit it to the sequence search tools of your choice. The format of a list file looks like this:
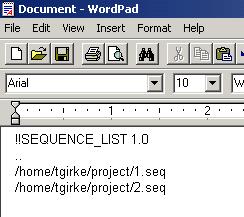
One
way of creating a list file is to select the sequences of your choice
in the Man List window and then save it as *.list under File
-> Save List As.
An alternative and often more flexible way of creating list files is to use a spread sheet program or WordPad on your local machine (use file extension *.list). To import a list file into the Main List, there are two options:
File -> Open List -> select *.list file
File -> Add Sequences From -> Sequence Files -> select *.list file
Note: List files with more than 2000 sequences cannot be expanded (viewed) in the Main List.
In addition to specifying query sequences, certain SeqLab application allow you to specify database records that will be used for a search or analysis. Programs that accept user-defined search sets are FastA, FindPatterns, FrameSearch, Overlap, ProfileSearch, SSearch and StringSearch. In all these programs you specify the search set by clicking on the Search Set button of the individual application, which opens a search set builder window. Note: Each application uses its own search set.
N. Creating Personal Sequence Databases with DataSet
To add your personal sequences to the Database Browser, you need to use the application DataSet. For this you first switch to the appropriate working directory (see C.), then you select your sequences or their list file in the Main List window, and choose: Functions -> Utilities -> Databases Utilities -> DataSet. You will be prompted with a dialog window where you assign a name and then press Run. This will add the following three files to your current working directory: *.header, *.ref and *.seq. When finished you should see your personal database in the Database Browser.
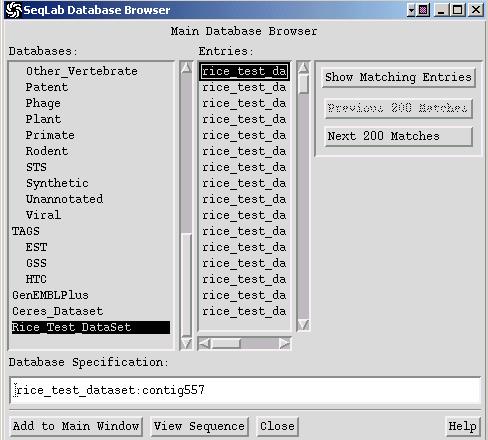
Note:
A DataSet is different from a BLASTable database, which
is explained in the next paragraph.
O. Creating BLASTable Sequence Databases
Create a new directory where you want to store your BLASTable databases and make it your working directory (see C.). Then you select the sequences that you wish to create a BLASTable set from, and choose: Functions -> Utilities -> Databases Utilities -> GCGtoBLAST.
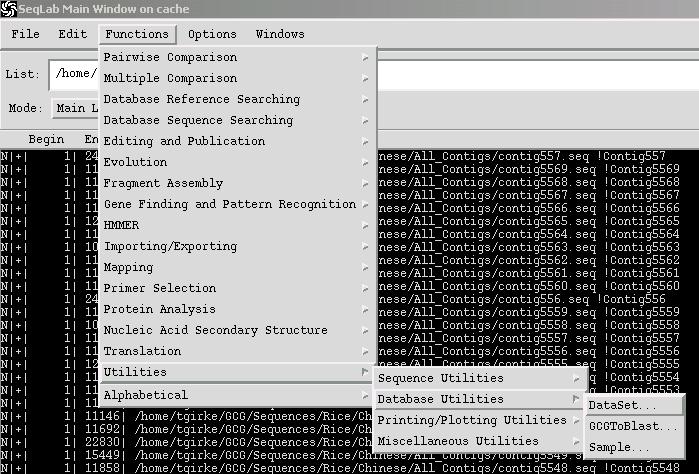
You will be presented with a dialog window that allows you to assign a name to the set. Enter a name and press Run. This operation creates five new files in your current working directory: *.phr, *.pin, *.psd, *.psi and *.psq. All sequence data are contained in this file structure. To save storage space, you can now delete the initial sequence files. Searching the database that you created requires that you first access the Wisconsin Package from the command line so that you can properly modify a configuration file, which is necessary to add a reference to your new BLASTable database to the BLAST database Search Set menu. To do this you would do the following from the UNIX command line after starting the Wisconsin Package there:
$ fetch genrundata:blast.sdbs
$ pico blast.sdbs (if you don't know how to use the pico editor you can update this text file in WordPad)
At the end of the file, add a line like: /path/db-base-name p my own blast database
Here are some notes for editing this line:
Substitute the actual full path to your newly created database for /path/
Substitute the base filename (the name you entered for the BLAST database when you created it) for db-base-name
The second column should be "p" for a protein database and "n" for a nucleotide database
To BLAST against your personal databases, your working directory needs to be the highest level in your home directory (this is a bug in our installation).
Within WinSCP: Create the following directories within the master directory Exercises: Seq, Pep, Database and Analysis. Use these directories to organize the work of the following exercises.
Within SeqLab: Create the same directory structure with the working director manager in SeqLab (see C.).
Import trace files: Download the trace files 09.ab1 & 13.ab1, import them into SeqLab, view trace plus text sequences, export the latter into FASTA or GenBank format and view them with WordPad on your local machine.
Import single sequences: Run in your web browser query "P450 & hydroxylase & acid & human [orgn]" against the NCBI Protein Database. Save the first ten proteins in FASTA and GenBank formats and import them one-by-one into SeqLab. Create alignment with Pileup.
Batch import: Import entire proteome of Halobacterium spec. from ftp://ftp.ncbi.nih.gov/genbank/genomes/Bacteria/Halobacterium_sp/AE004437.faa.
Import alignments: Create multiple alignment of sequences from 2.2. using MultAlin. Import alignment in MSF and FASTA formats.
Export: Export in single and batch sequence modes. Export alignment in MSF format.
Features
In sequence: view imported sequence from 2.2. in Editor, display and add features.
In alignment: run Pileup with Lookup list file from 4.1. and transfer alignment annotations into Editor and find heme binding cystein residue, export alignment and view it in GeneDoc (only on PC).
Database searches: Lookup, FASTA, SSearch, BLAST, HMMER
Lookup: run query "CYPIII (All text) & P450 (Def)" in Lookup against SwissProt database.
SSearch, FASTA, BLAST and PSI-BLAST: query with one of these sequences the SwissProt database using SSearch, BLAST and FASTA.
HMMER: Align sequences from 4.1. Retrieve and align remote homologs from SwissProt database with HMMER: HmmerBuild, HmmerCalibrate, HmmerSearch and HmmerAlign.
Create BLASTable database
Create BLASTable database for proteome from Halobacterium spec. (imported under 2.3.).
Pattern Searches
Motifs: Use Motifs to find PROSITE patterns in protein alignment from 2.2., find pattern with Edit/Find and highlight it in all sequences at once using the Feature function.
FindPattern: find out how many sequences in the SwissProt database share this pattern using FindPattern.
Consensus and FitConsensus: retrieve the corresponding nucleotide sequences, align them, calculate consensus sequence with Consensus and query with it a small nucleotide database using FitConsensus.
MEME and MotifSearch: use MEME to find conserved motifs in your set of unaligned nucleotide sequences. Use the resulting MEME profiles to query a small nucleotide database with MotifSearch.
Phylogenetic Trees:
PAUP: use PaupSearch to generate a bootstrapped tree from alignment under 2.2. Edit tree with PaupDisplay, Treeview (local) and PowerPoint.
Distance Matrix: calculate distance matrix for alignment using Distances and plot its tree with Display.
Molecular tools: Primer design, backtranslate
Primer design: Design primers that amplify the longest ORFs of the two sequences from 2.1.
Restriction map: generate restriction map for one of the sequences from 1.1. using Map.